Markergene in Zellclustern finden
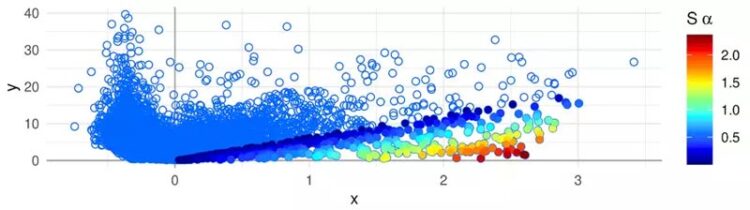
Association Plot eines Zellclusters, der mit dem APL-Paket erstellt wurde - die Gene sind als Punkte dargestellt. Die Farbskala auf der rechten Seite zeigt die Stärke der Assoziation der Gene zum Cluster: Rot gefärbte Gene sind am stärksten clusterspezifisch.
© E. Gralinska, MPIMG
Neue Methode erleichtert die Identifikation von Zelltyp-spezifischen Genen in Single-Cell-Daten: Die abertausenden Zellen in einer biologischen Probe sind alle individuell unterschiedlich und lassen sich einzeln analysieren. Anhand der Gene, die in ihnen aktiv sind, lassen sie sich in „Cluster“ zusammen sortieren. Aber welche Gene sind besonders charakteristisch für Cluster, was sind also ihre „Markergene“? Ein neues bioinformatisches Verfahren namens Association Plot erleichtert die Analyse dieser Daten.
Welche Gene sind spezifisch für einen bestimmten Zelltyp, „markieren“ also deren Identität? Wegen immer größer werdender Datenmengen wird diese Frage immer schwieriger zu beantworten. Häufig sind Markergene einfach Gene, die über Jahre hinweg immer wieder in bestimmten Zellpopulationen gefunden wurden. Jedoch könnten noch viel mehr Gene für einen bestimmten Zelltyp charakteristisch sein, die bisher noch unentdeckt sind.
Ein neues statistisches Verfahren zur Visualisierung der Genaktivität innerhalb eines Zellclusters erleichtert es, dessen Markergene zu finden. Diese „Association Plots“ (APL) vergleichen die Gene eines Clusters mit allen anderen Clustern des Datensatzes. Auch welche Gene in anderen Clustern vorkommen, lässt sich im APL-Diagramm leicht ablesen.
„Mit APL lassen sich nicht nur neue Markergene identifizieren, es funktioniert auch umgekehrt. In einem Datensatz mit unbenannten Clustern können wir Zelltypen bestimmen, wenn wir eine Liste bekannter Markergene als Grundlage nehmen“, sagt Elzbieta Gralinska vom Max-Planck-Institut für molekulare Genetik.
Die Biotechnologin arbeitet im Team von Martin Vingron, welches APL entwickelt, seine Funktion an zwei öffentlich verfügbaren Datensätzen demonstriert und die Ergebnisse in der Fachzeitschrift Journal of Molecular Biology veröffentlicht hat. Zudem ist APL als kostenloses Modul für die Statistik-Umgebung R erschienen. Das APL-Modul erlaubt es den Forschenden, ihre Single-Cell-Daten visuell zu inspizieren und für detaillierte Einzelheiten einzelne Datenpunkte mit der Computermaus auszuwählen.
Einzelne Zellen analysieren und gruppieren
Warum ist es überhaupt notwendig, Markergene zu ermitteln? Moderne Sequenziertechnologien können inzwischen einzelne Erbgut-Moleküle in einzelnen Zellen analysieren. So kann etwa aus einer Blutprobe jede Zelle vereinzelt und eine Stichprobe der enthaltenen RNA entschlüsselt werden. Diese Daten repräsentieren aktive Gene, die zu RNA-Molekülen transkribiert wurden.
Der Vorteil: Statt zu rätseln, aus welchem Zelltyp nun eine bestimmte RNA stammt, lässt sich diese zu seinem Ursprung zurückverfolgen. Der Nachteil: Sequenzieren die Forschenden tausende RNA-Transkripte in jeder einzelnen von zehntausenden Zellen, entstehen schnell unübersichtliche Datenberge.
Ein Ausweg ist, die Zellen anhand ihrer Eigenschaften zu sortieren. „Einzelzelldaten setzen sich aus Vertretern verschiedenster Zelltypen zusammen. Wir sind jeweils an Zellen desselben Zelltyps interessiert, die sich alle ähnlich verhalten sollten“, erklärt Martin Vingron. Daher sei es sinnvoll, ähnliche Zellen vom Computer zu Gruppen zusammenfassen zu lassen, sagt er. „Für uns werden Zelltypen durch ihre Markergene definiert.“
Interaktiv Cluster erforschen
Anhand öffentlich verfügbarer Daten von weißen Blutzellen demonstrierte das Team sein neues Verfahren. Die vielen verschiedenartigen weißen Blutkörperchen wie T-Zellen, B-Zellen oder Monozyten befinden sich in unterschiedlichen Clustern. Die Forschenden bestätigten bekannte Markergene und konnten zeigen, dass enge Verwandte in der Gruppe der weißen Blutzellen auch große Ähnlichkeit in ihrer Genaktivität aufweisen.
„Jedes der charakteristischen Gene, die wir mit APL gefunden haben, wird von mindestens einer anderen Methode zum Aufspüren dieser Gene gefunden“, sagt Gralinska. Denn zur Bestimmung von Markergenen in Clustern existieren zwar bereits Algorithmen, erklärt die Forscherin. Doch die grafische Darstellung der Ergebnisse als Association Plots sei äußerst vorteilhaft. „Bestehende Verfahren liefern lediglich lange Listen von Genen und Score-Werten. User gehen die Liste häufig durch und brechen dann bei einem willkürlichen Schwellenwert ab“, sagt Gralinska.
Die neue Methode dagegen biete eine Möglichkeit, diese Gene zu visualisieren, auf jedes einzelne Gen zu klicken und dessen Aktivität genauer unter die Lupe zu nehmen. „Wir stellen nicht nur Listen von Markergenen zur Verfügung, sondern die Benutzerinnen und Benutzer können auch überprüfen, wie sich diese Gene verhalten“, sagt die Forscherin. „Mit Association Plots können sie in ihre Daten eintauchen, um mehr über die einzelnen Zelltypen zu erfahren.“ Zudem sei es sehr einfach, über kompatible Software in einem weiteren Schritt eine Gene-Ontology-Enrichment-Analyse durchzuführen. Dadurch ließe sich die biologische Funktion der interessantesten Gene aufschlüsseln – „ein sehr nützliches Feature“, findet Gralinska.
Das zugrundeliegende mathematische Modell
Die hochdimensionalen Daten aus Genaktivitäten von Zellen lassen sich visuell nicht ohne Informationsverlust darstellen. Dies erschwert auch die Analyse von Clusterdaten. „Unser Trick ist, dass wir viel mehr als nur zwei oder drei Dimensionen einbeziehen, letztlich aber ein zweidimensionales Diagramm erstellen können“, sagt Gralinska.
Den Association Plots liegt ein mathematisches Verfahren zugrunde, das Gene und Zellen in einem hochdimensionalen Raum einbettet. Durch die Messung der Abstände zwischen Genen und Zellen in diesem Raum ergeben sich Wertepaare, die einerseits die Verbundenheit eines Gens zum eigenen Cluster und andererseits die Assoziation zu den anderen Clustern widerspiegeln.
„Ein Nachteil der Association Plots ist, dass wir auf geclusterte Daten angewiesen sind. Für das Clustering müssen wir andere Techniken einsetzen“, sagt Martin Vingron. „Nichtsdestotrotz hoffen wir, dass unser neues Verfahren viele neue Anwenderinnen und Anwender findet. Wir finden, dass ein visueller und interaktiver Prozess die Analyse einfach besser macht.“
Wissenschaftliche Ansprechpartner:
Prof. Martin Vingron
Direktor, Leiter der Abteilung „Bioinformatik“
Max-Planck-Institut für Molekulare Genetik
+49 30 8413-1150
vingron@molgen.mpg.de
Elzbieta Gralinska
Wissenschaftlerin
Max-Planck-Institut für Molekulare Genetik
+49 30 8413-1173
gralinska@molgen.mpg.de
Originalpublikation:
Gralinska E, Kohl C, Fadakar BS, Vingron M. Visualizing cluster-specific genes from single-cell transcriptomics data using association plots. J Mol Biol. doi:10.1016/j.jmb.2022.167525
Weitere Informationen:
https://www.molgen.mpg.de/4521121/ – Webversion dieser Pressemitteilung
https://www.sciencedirect.com/science/article/pii/S0022283622000997 – Originalpublikation
https://vingronlab.github.io/APL/ – Website des APL-Programms
Media Contact
Alle Nachrichten aus der Kategorie: Biowissenschaften Chemie
Der innovations-report bietet im Bereich der "Life Sciences" Berichte und Artikel über Anwendungen und wissenschaftliche Erkenntnisse der modernen Biologie, der Chemie und der Humanmedizin.
Unter anderem finden Sie Wissenswertes aus den Teilbereichen: Bakteriologie, Biochemie, Bionik, Bioinformatik, Biophysik, Biotechnologie, Genetik, Geobotanik, Humanbiologie, Meeresbiologie, Mikrobiologie, Molekularbiologie, Zellbiologie, Zoologie, Bioanorganische Chemie, Mikrochemie und Umweltchemie.


Neueste Beiträge

Neuartiges Material für nachhaltiges Bauen
Innovativer Werkstoff für eine energieeffiziente Architektur: Forschende des Karlsruher Instituts für Technologie (KIT) stellen in der aktuellen Ausgabe der Fachzeitschrift Nature Communications ein polymerbasiertes Material mit besonderen Eigenschaften vor. Das…

Neues Antibiotikum gegen Erreger der Flussblindheit und Lymphatischen Filariose
Prof. Achim Hoerauf, Direktor des Instituts für Medizinische Mikrobiologie, Immunologie und Parasitologie des Universitätsklinikums Bonn (UKB), und seinem Team ist es in Kollaboration mit der Abteilung Pharmazeutische Technologie und Biopharmazie…
Evolutionäre Genomik: Folgen biodiverser Fortpflanzungssysteme
Die Deutsche Forschungsgemeinschaft (DFG) fördert die Einrichtung eines neuen Graduiertenkollegs (GRK) in der Biologie an der Universität Göttingen. Das GRK mit dem Titel „Evolutionary Genomics: Consequences of Biodiverse Reproductive Systems…

