Chemie im Computer: neuartige Computersimulationen chemischer Reaktionen in der Halbleiterindustrie
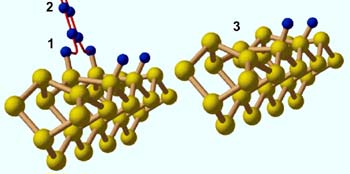
Quanten-Monte-Carlo Rechnungen erklären die Ablösung (Desorption) von molekularem Wasserstoff von einer Siliziumoberfläche. Das Bild links zeigt, wie diese Reaktion auf atomarer Ebene abläuft: Zwei Wasserstoffatome (blau), lösen sich von den Siliziumatomen der Oberfläche (gelb) ab (als 1 markiert), nähern sich entlang ihres Reaktionsweges an (rot gezeichnet) und verbinden sich schließlich zu einem Wasserstoffmolekül (als 2 markiert), das in die Gasphase desorbiert. Das Bild rechts zeigt die Siliziumoberfläche nach der Desorption: Ein Siliziumatom, an das zuvor ein Wasserstoffatom gebunden war, hat nun eine andere, energetisch günstigere Position eingenommen und ist in die Oberfläche hinein gewandert (als 3 markiert)
Forscherteam um Berliner Max-Planck-Wissenschaftler gelingt mit neuartigen Computersimulationen die Beschreibung einer wichtigen chemischen Reaktion in der Halbleiterindustrie
Viele Eigenschaften von Halbleitern werden gezielt durch chemische Reaktionen an ihren Oberflächen manipuliert. Doch wie diese Prozesse auf atomarer Ebene ablaufen, ist noch weitestgehend unbekannt. Jetzt haben Forscher des Fritz-Haber-Instituts in Berlin sowie der Universitäten Essen, Leiden (Niederlande) und Cork (Irland) mit Hilfe von Computersimulationen geklärt, in welchen Einzelschritten ein solcher technologisch wichtiger Prozess – die Reaktion von Wasserstoff mit Silizium – tatsächlich verläuft (Physical Review Letters, 14. Oktober 2002). Dazu setzten die Forscher erstmals ein so genanntes Quanten-Monte-Carlo-Verfahren ein, mit dem sich Reaktionsschritte weit genauer als bisher berechnen lassen: Damit können Prozesse im Computer in Ruhe beobachtet werden, die in realen chemischen Reaktionen nur wenige Nanosekunden dauern und auf einer Fläche von Millionstel Millimetern stattfinden.
Ob in der Forschung oder der industriellen Fertigung, Halbleitermaterialien mit gewünschten Eigenschaften entstehen meist über eine Kette von chemischen Reaktionen. Um diese besser steuern zu können, arbeiten Forscher heute intensiv daran, die einzelnen Schritte solcher Reaktionen im Detail, sozusagen aus der Sicht der beteiligten Atome, zu verstehen. Dazu greifen Wissenschaftler immer häufiger auf den Computer zurück und simulieren das Verhalten der Moleküle. Bisher gelang das für die an sich einfache und gut untersuchte Reaktion von Wasserstoff mit Siliziumoberflächen nur sehr unvollständig. Diese Reaktion ist technologisch bedeutsam, etwa für das Ätzen und Oberflächenveredeln oder das Wachsen von Siliziumkristallen.
Die bisher zur Simulation dieser Reaktion eingesetzten Modelle konnten die experimentellen Befunde nicht schlüssig erklären. Vielmehr sehen sich die Forscher mit dem berüchtigten „Rätsel der Energiebarrieren“ konfrontiert: Wasserstoff-Moleküle, die auf die Silizium-Oberfläche auftreffen, müssen zunächst in einzelne Wasserstoffatome zerfallen (dissoziieren), die dann wiederum chemische Bindungen mit Silizium-Atomen eingehen (adsorbieren). Auf dem Weg dahin müssen die Moleküle eine hohe Energiebarriere überwinden. Deshalb dissoziieren aus einem Wasserstoffgas bei Raumtemperatur nur jene Moleküle, die zufällig eine hohe Energie haben, tatsächlich an der Oberfläche. Erstaunlicherweise zeigen die Moleküle jedoch beim umgekehrten Prozess, der Desorption von der Oberfläche, im Experiment keine besonders hohe Energie. Deshalb scheint es, als könnten sie die Oberfläche auf einem ganz anderen Weg verlassen, ohne eine Energiebarriere überqueren zu müssen.
Theoretische Modelle können diese unterschiedlichen Reaktionswege zwar nachbilden, stehen bislang aber auf recht wackligen Beinen: den dazu durchgeführten Dichtefunktional-Berechnungen hapert es an Genauigkeit, da sie die Wechselwirkungen der Elektronen nur näherungsweise erfassen. Auch Referenzrechnungen mit genaueren quanten-chemischen Methoden helfen nicht weiter. Wegen des dazu erforderlichen hohen Rechenaufwandes können sie nur kleinere Modellsysteme mit wenigen Dutzend Atomen behandeln, so dass wichtige strukturelle Aspekte der realen Siliziumoberfläche außer Acht bleiben.
Die Berliner Max-Planck-Forscher und ihre Kollegen von den Universitäten Essen, Leiden (NL) und Cork (IR) haben nun zur Berechnung der einzelnen Reaktionsschritte einen neuen theoretischen Ansatz gewählt, der die Nachteile herkömmlicher Rechenverfahren überwindet. Dazu verwendeten sie die so genannte (diffusive) Quanten-Monte-Carlo-Methode, mit der die chemischen Bindungen in einem realistischen Reaktionskomplex weitgehend näherungsfrei errechnet werden können. Das Verfahren ist den heute gebräuchlichen Dichtefunktional-Methoden an Genauigkeit überlegen. Darüber hinaus ist es auch auf Systeme mit mehr als hundert Atomen anwendbar, die mit genaueren Methoden aus der Quanten-Chemie rechnerisch nicht mehr zu bewältigen wären.
Den Berliner Wissenschaftlern und ihren Kollegen gelang es mit der neuen Methode, die bisher widersprüchlichen theoretischen und experimentellen Befunde zu dieser Reaktion miteinander in Einklang zu bringen. „Wir bestimmen damit die Struktur und Energie der Moleküle – sprich, ihr chemisches Verhalten – allein an Hand der Ordnungszahlen der beteiligten Atome. Die Quanten-Monte-Carlo-Methode stützt sich dabei auf keinerlei empirische Parameter, sie ist, kurz gesagt, eine ab-initio-Methode,“ erläutert dazu Peter Kratzer, Wissenschaftler am Berliner Fritz-Haber-Institut. Im Grunde möchten die Forscher in einem Molekül die Verteilung der Elektronen bestimmen, die sich nach den Regeln der Quantenmechanik aus der Lösung einer Vielteilchen-Schrödinger-Gleichung ergibt. Die Quantennatur und gegenseitige Abstoßung der Elektronen macht dies jedoch zu einer äußerst komplexen Aufgabe: Beides führt dazu, dass die Bewegungen aller Elektronen nur zusammen, also in Korrelation zueinander, und nicht unabhängig voneinander beschrieben werden können.
Das Quanten-Monte-Carlo Verfahren löst nun die Schrödinger-Gleichung mit einem statistischen Algorithmus. Die hochdimensionale elektronische Vielteilchen-Wellenfunktion wird dabei, ausgehend von einer geeigneten Versuchswellenfunktion, direkt simuliert. Die statistische Genauigkeit lässt sich hierbei – über die Einbeziehung von immer mehr elektronischen Konfigurationen in die Simulation – schrittweise erhöhen. Im Ergebnis können auf diese Weise molekulare Energien sehr genau abgeschätzt werden. Ein weiterer entscheidender Vorzug der Quanten-Monte-Carlo-Verfahren für die tägliche Rechenpraxis ist, dass sie sich vergleichsweise leicht auf Parallelrechnern umsetzen lassen und so die Rechenzeit stark verkürzt werden kann.
Mit ihren Berechnungen konnten die Wissenschaftler nun das „Barrieren-Rätsel“ bei dieser Reaktion in seinem Kern zweifelsfrei aufklären. Danach müssen die Wasserstoffmoleküle bei der Adsorption auf einer reinen Siliziumoberfläche tatsächlich eine sehr hohe Energiebarriere überwinden. Umgekehrt verläuft die Desorption von der Oberfläche, die weitgehend mit Wasserstoffatomen bedeckt ist, ganz anders ab: Wenn sich zwei Atome zu einem Wasserstoffmolekül vereinigen, durchlaufen sie einen Reaktionsweg, der ein eher flaches Energieprofil aufweist. Dieses Szenario steht im Einklang mit experimentellen Analysen und bildet die Basis für weiterführende Untersuchungen, die den Einfluss von Oberflächendefekten (wie Stufen und Kanten) auf das Reaktionsgeschehen unter die Lupe nehmen.
Matthias Scheffler, Direktor der Abteilung Theorie am Fritz-Haber-Institut, erklärt dazu: „Das Quanten-Monte-Carlo-Verfahren hat sich als sehr nützlich für unser Verständnis erwiesen, und zwar bei einer Reaktion, bei der traditionelle Ansätze überfordert waren. Ich denke, dieses Verfahren kann unser methodisches Instrumentarium sehr sinnvoll ergänzen.“ Bei aller Zuversicht, so Scheffler weiter, „muss es als relativ junges Rechenverfahren noch erheblich weiter entwickelt werden, um ein ähnlich vielseitiges Instrument zu werden wie es heute etwa die Dichtefunktional-Verfahren sind, die sich bis in die industrielle Forschung hinein durchgesetzt haben.“
Dr. Peter Kratzer
Fritz-Haber-Institut der Max-Planck-Gesellschaft
Faradayweg 4-6
14195 Berlin
Tel.: 030 – 8413 – 4809
Fax: 030 – 8413 – 4701
E-Mail: kratzer@fhi-berlin.mpg.de
Media Contact
Weitere Informationen:
http://www.mpg.de/pri02/pri02100.htm http://www.fhi-berlin.mpg.de http://www.mpg.de/pri02/pri02100.pdfAlle Nachrichten aus der Kategorie: Verfahrenstechnologie
Dieses Fachgebiet umfasst wissenschaftliche Verfahren zur Änderung von Stoffeigenschaften (Zerkleinern, Kühlen, etc.), Stoffzusammensetzungen (Filtration, Destillation, etc.) und Stoffarten (Oxidation, Hydrierung, etc.).
Unter anderem finden Sie Wissenswertes aus den Teilbereichen: Trenntechnologie, Lasertechnologie, Messtechnik, Robotertechnik, Prüftechnik, Beschichtungsverfahren und Analyseverfahren.




Neueste Beiträge

Ideen für die Zukunft
TU Berlin präsentiert sich vom 22. bis 26. April 2024 mit neun Projekten auf der Hannover Messe 2024. Die HANNOVER MESSE gilt als die Weltleitmesse der Industrie. Ihr diesjähriger Schwerpunkt…

Peptide auf interstellarem Eis
Dass einfache Peptide auf kosmischen Staubkörnern entstehen können, wurde vom Forschungsteam um Dr. Serge Krasnokutski vom Astrophysikalischen Labor des Max-Planck-Instituts für Astronomie an der Universität Jena bereits gezeigt. Bisher ging…

Wasserstoff-Produktion in der heimischen Garage
Forschungsteam der Frankfurt UAS entwickelt Prototyp für Privathaushalte: Förderzusage vom Land Hessen für 2. Projektphase. Wasserstoff als Energieträger der Zukunft ist nicht frei verfügbar, sondern muss aufwendig hergestellt werden. Das…

