Forscher entdecken Enzym-Erbkrankheit
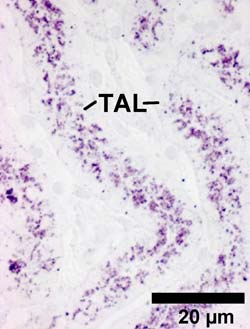
Ausbruch der „Dierks'schen Krankheit“: Die mikroskopische Aufnahme zeigt starke Anreiche-rungen von Heparansulfat (blau-rote Färbung) in Zellen vom Mittelstück der Nierenkanälchen (TAL). Schuld ist das fehlerhafte Enzym ARSG. Foto: Universität Bielefeld<br>
Ein internationales Forscherteam unter Leitung von Professor Dr. Thomas Dierks von der Universität Bielefeld hat eine Enzym-Erbkrankheit entdeckt. Sie ist eine Unterform des Mucopolysaccharidose-Syndroms und trägt den Namen MPS IIIE oder – nach dem Entdecker – „Dierks‘sche Krankheit“.
Ihre Folgen zeigen sich bei Mäusen im fortschreitenden Verlust der geistigen Fähigkeiten, vor allem Vergesslichkeit, Lern- und Koordinations-schwierigkeiten. Der Biochemiker Dierks und sein Team haben nicht nur die Krankheit identifiziert – sie haben auch ein Therapiekonzept entwickelt. Ihre Ergebnisse präsentieren sie in dieser Woche (KW 24) in der angesehenen Fachzeitschrift „Proceedings of the National Academy of Sciences of the USA“ (PNAS).
Enzyme steuern den Bau und die Aufspaltung von Nähr- und Botenstoffen im menschlichen Körper. Wenn der Körper wegen einer vererbten Störung ein fehlerhaftes Enzym produziert, dann versagt diese Steuerung: Der Mensch wird krank, zum Beispiel weil sich in seinem Körper Stoffe anreichern, die nicht mehr gespalten werden können.
Dierks und seine Kollegen haben herausgefunden, dass eine Schädigung des Enzyms Arylsulfatase G (ARSG) in Mäusen die Krankheit MPS IIIE auslöst. Eigentlich ist das Enzym mit dafür zuständig, das Kohlenhydrat Heparansulfat aufzuspalten. Das geschieht innerhalb der Zellen – in den Lysosomen. In diesen „Recyclinganlagen“ der Zellen werden nicht mehr benötigte Heparansulfat-Moleküle in ihre kleinsten Bausteine zerlegt, aus denen später wieder neue Moleküle zusammengesetzt werden.
Das Heparansulfat ist ein langkettiges Molekül. Diese Kette kann immer nur von einem Ende her zerlegt werden und immer nur Stück für Stück. Das geschieht durch verschiedene Enzyme, zu denen ARSG gehört. Wenn eins von den Enzymen aufgrund eines genetischen Defekts aus-fällt, bricht die gesamte Zersetzung ab. Die Molekülketten bleiben liegen und reichern sich im Lysosom immer weiter an, das schließlich aufhört, zu arbeiten. Dann werden auch andere Stoffe, wie zum Beispiel Proteine und Fette, nicht mehr abgebaut und sammeln sich an. Das Lysosom dehnt sich immer weiter aus, bis es die gesamte Zelle schädigt und schließlich unter-gehen lässt.
Zu Beginn der Studie (2003) waren sieben der Enzyme bekannt, die am Abbau von Heparansulfat beteiligt sind. Dierks und sein Team haben gezielt nach einem fehlenden Enzym gesucht. Sie wussten, dass insgesamt mindestens neun verschiedene Enzyme für den vollständigen Abbau des Heparansulfats zuständig sein müssen – zwei von ihnen, beides Sulfatasen, blieben bis dahin unentdeckt. Jede vererbte Störung von einem dieser Enzyme entspricht jeweils einer Krankheit, die zum Mucopolysaccharidose-Syndrom zählt. „Als wir mit der Untersuchung anfingen, vermuteten wir, dass die Arylsulfatase G mit dem Abbau des Heparansulfats zu tun hat“, sagt Dierks. Um die Annahme zu testen, erzeugte sein Team Mäu-se, in denen die Arylsulfatase G defekt war. Die Vermutung bewahrheitete sich. In den Tieren fanden sich mit zunehmendem Alter hohe Konzentrationen von Heparansulfat, und zwar im Gehirn, in der Leber und in den Nieren. Professor Jeffrey D. Esko von der University of California San Diego (USA) arbeitet in dem Projekt mit. Seine Mitarbeiter und er untersuchten Gewebeproben mit einem Massenspektrometer und bestätigten das Ergebnis, wonach die blockierte Abspaltung einer bestimmten Sulfatgruppe die Ursache der Heparansulfat-Ansammlung ist.
Mit Verhaltenstests fand das Team von Dierks heraus, dass Mäuse mit ARSG-Mangel ab einem Alter von zwölf Monaten mit kognitiven Störungen zu kämpfen haben: Wenn sie auf ein freies Feld kommen, bleiben sie im Gegensatz zu ihren gesunden Artgenossen am sicheren Rand und trauen sich nicht, den Platz zu erkunden. Auch beim Wasserlabyrinth-Test scheitern sie: Lange hatten die Mäuse erfolgreich trainiert, in einem Pool, der mit milchiger Flüssigkeit gefüllt ist, schwimmend eine unter der Oberfläche versteckte Plattform zu finden – doch sobald sie zwölf Monate alt waren, konnten sie sich nicht mehr die Position der Plattform merken. Sie brauchten deutlich länger als zuvor, um sie zu entdecken. Jüngere und gesunde Mäuse fanden die Plattform problemlos wieder. Der Defekt liegt im Gehirn. Mit einer Untersuchung von Gewebeproben aus dem Kleinhirn der Tiere zeigte das Forscherteam, dass durch die Anhäufung des Heparansulfats die Purkinje-Zellen im Kleinhirn absterben und, begleitet von Entzündungen, durch neue Zellen ersetzt werden. Diese Glia-Zellen haben laut Dierks aber nur noch eine Stützfunktion und bilden keine neuen Nervenverbindungen aus.
Der große Erfolg der Forscher: Durch ihre Erkenntnisse lässt sich eine Therapie für die Erbkrankheit entwickeln und an den Mäusen testen. Sie stellen das ARSG-Enzym künstlich her – in einem biotechnologischen Verfahren mit Hilfe gentechnisch veränderter Zellkulturen. Bei erkrankten Mäusen, denen regelmäßig eine Lösung mit dem Enzym gespritzt wird, sollten die Schädigungen der Organe gestoppt werden. Ähnliche Behandlungen waren laut Dierks auch bei Patienten mit anderen Mucopolysaccharidose-Erkrankungen erfolgreich. „Die biochemischen Abläufe, die mit solchen lysosomalen Speicherkrankheiten zusammenhängen, sind bei allen Säugetieren prinzipiell gleich, wirken sich aber beim Menschen aufgrund des höheren Lebensalters schwerwiegender aus“, so Dierks. Manchmal sei es schwer, diese Krankheiten rechtzeitig zu diagnostizieren, weil sie oft schleichend einsetzen. Mitunter lässt sich die Krankheit an ihren Symptomen erst im Jugendalter erkennen. Und dann ist die Diagnose für Ärzte schwierig, sagt Thomas Dierks, „weil systematische Untersuchungen nur im Kindesalter durchgeführt werden und man zunächst einmal nicht an eine erbliche Ursache denkt. Die Therapie muss aber so früh wie möglich beginnen“. Die „Dierks‘sche Krankheit“ kann durch eine Untersuchung im Massenspektrometer eindeutig diagnostiziert werden – die Forscher haben dafür ein eigenes Verfahren entwickelt.
Zum Forschungsteam von Thomas Dierks gehören außer Wissenschaftlerinnen und Wissenschaftlern der Universität Bielefeld auch Kollegen der Katholieke Universiteit Leuven, Belgien, der University of California San Diego, USA, sowie der Georg-August Universität Göttingen und der Christian-Albrechts-Universität zu Kiel.
Originalveröffentlichung:
Arylsulfatase G Inactivation Causes Loss of Heparan Sulfate 3-O-Sulfatase Activity and Mucopo-lysaccharidosis in Mice, Björn Kowalewski, William C. Lamanna, Roger Lawrence, Markus Damme, Stijn Stroobants, Michael Padva, Ina Kalus, Marc-André Frese, Torben Lübke, Renate Lüllmann-Rauch, Rudi D’Hooge, Jeffrey D. Esko, Thomas Dierks, Proceedings of the National Academy of Sciences of the USA, Juni 2012, dx.doi.org/10.1073/pnas.1202071109.
Kontakt:
Prof. Dr. Thomas Dierks, Universität Bielefeld
Fakultät für Chemie, Arbeitsgruppe Biochemie I
Telefon: 0521 106-2092
E-Mail: thomas.dierks@uni-bielefeld.de
Media Contact
Weitere Informationen:
http://www.uni-bielefeld.deAlle Nachrichten aus der Kategorie: Biowissenschaften Chemie
Der innovations-report bietet im Bereich der "Life Sciences" Berichte und Artikel über Anwendungen und wissenschaftliche Erkenntnisse der modernen Biologie, der Chemie und der Humanmedizin.
Unter anderem finden Sie Wissenswertes aus den Teilbereichen: Bakteriologie, Biochemie, Bionik, Bioinformatik, Biophysik, Biotechnologie, Genetik, Geobotanik, Humanbiologie, Meeresbiologie, Mikrobiologie, Molekularbiologie, Zellbiologie, Zoologie, Bioanorganische Chemie, Mikrochemie und Umweltchemie.




Neueste Beiträge

Immunzellen in den Startlöchern: „Allzeit bereit“ ist harte Arbeit
Wenn Krankheitserreger in den Körper eindringen, muss das Immunsystem sofort reagieren und eine Infektion verhindern oder eindämmen. Doch wie halten sich unsere Abwehrzellen bereit, wenn kein Angreifer in Sicht ist?…
Durchbruch bei CRISPR/Cas
Optimierte Genschere erlaubt den stabilen Einbau von großen Genen. Großer Fortschritt an der CRISPR-Front. Wissenschaftlern des Leibniz-Instituts für Pflanzenbiochemie (IPB) ist es erstmals gelungen, sehr effizient große Gen-Abschnitte stabil und…

Rittal TX Colo: Das neue Rack für Colocation Data Center
Rittal TX Colo: Flexibel, skalierbar und zukunftssicher Mit der zunehmenden Digitalisierung und künftig auch immer mehr KI-Anwendungen steigt der Bedarf an Rechenleistung signifikant – und damit boomt der Colocation-Markt. Unternehmen…

