Lichtblicke für die Therapie des humanen Usher-Syndroms
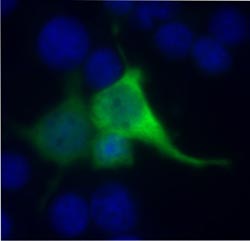
Fluoreszenzmikroskopische Analyse von Zellen mit einer Nonsens-Mutation im Usher-Syndrom-1C-Gen (USH1C/Harmonin). Nach Applikation des Designer-Aminoglykosids NB54 wird die USH1C-Mutation überlesen und die therapierten Zellen produzieren das gesunde Harmonin-Protein (grün).<br><br>Quelle: K. Nagel-Wolfrum, Institut für Zoologie, JGU<br>
Nach langjähriger Grundlagenforschung erkennen Wissenschaftler der Johannes Gutenberg-Universität Mainz (JGU) immer besser die Hintergründe des humanen Usher-Syndroms und kommen möglichen Therapieansätzen auf die Spur.
Die Wissenschaftler aus der Usher-Forschergruppe um Univ.-Prof. Dr. Uwe Wolfrum testen dabei vor allem zwei Wege, um entweder mutierte Gene zu reparieren oder aber den Gendefekt durch Einsatz von Wirkstoffen auszuschalten. Den bisherigen Ergebnissen zufolge erscheinen beide Optionen vielversprechend. Das Usher-Syndrom ist eine vererbte Erkrankung, bei der gleichzeitig eine Hör- und Sehbehinderung auftritt.
Das Usher-Syndrom ist mit einer Häufigkeit von 1:6.000 die häufigste Form angeborener Taub-Blindheit des Menschen. Für die Betroffenen bedeutet diese Krankheit eine große Einschränkung in ihrem alltäglichen Leben, weil die beiden wichtigsten Sinnesorgane Ohr und Auge betroffen sind. Im schwerwiegendsten Fall werden die Patienten taub geboren und leiden bereits vor der Pubertät an Sehstörungen, die sich in einer Degeneration der Netzhaut manifestieren und zur völligen Erblindung führen. Während der Gehörverlust mit Hörgeräten und Cochlea-Implantaten kompensiert werden kann, gibt es bislang noch keine Therapiemöglichkeit für das Auge. Wissenschaftler der JGU haben nun in aktuellen translationalen präklinischen Studien Therapieansätze für die Krankheit untersucht.
Bei seinen Untersuchungen konzentrierte sich das Mainzer Usher-Therapieteam um Dr. Kerstin Nagel-Wolfrum auf eine Nonsense-Mutation im USH1C-Gen, die bei einer deutschen Familie zu der schwerwiegendsten Form des Usher-Syndroms führt. Bei der Nonsense-Mutation entsteht in der DNA ein Stopp-Signal und es kommt folglich zu einem vorzeitigen Abbruch der Synthese des Harmonin-Proteins, das vom USH1C-Gen kodiert wird.
Das Forscherteam konnte seine neuesten Ergebnisse zur Genreparatur als Therapiemöglichkeit für die Behandlung von Usher-Syndrom-Patienten in der Juni-Ausgabe der ophthalmologischen Fachzeitschrift „Investigative Ophthalmology & Visual Science“ publizieren. Es war Dr. Nora Overlack im Rahmen ihrer Doktorarbeit gelungen, das USH1C-Gen mit molekularen Scheren, eigens generierten sogenannten Zinkfingernukleasen, zu reparieren. Die Wissenschaftler haben zunächst durch die Zinkfingernukleasen einen Doppelstrangbruch in der DNA im Bereich der krankheitsverursachenden Mutation ausgelöst. Dieser molekulare chirurgische Schnitt wurde mithilfe des zelleigenen Reparaturmechanismus der homologen Rekombination und eines parallel eingeführten, nicht-mutierten USH1C-DNA-Strangs repariert. Dadurch wurde die mutierte Gensequenz gegen die nicht-mutierte Sequenz ausgetauscht. Die Funktionalität der Zinkfingernukleasen konnte im Zellkulturmodellsystem sowohl auf genomischer Ebene als auch auf Proteinebene für die erfolgreiche Genreparatur demonstriert werden.
Darüber hinaus publizierte das Forscherteam gerade seine neuesten Arbeiten zu den pharmakogenetischen Therapieansätzen für die Behandlung von Usher-Syndrom-Patienten mit Nonsense-Mutationen in der Fachzeitschrift „EMBO Molecular Medicine“. In dieser Arbeit vergleichen Dr. Tobias Goldmann und Teamkollegen verschiedene Moleküle, die das Überlesen des Stopp-Signals induzieren und so eine normale Proteinsynthese sicherstellen. Darüber hinaus evaluieren sie die Biokompatibilität der unterschiedlichen Moleküle in der Retina. Dabei standen PTC124 (Ataluren®) und Designer-Aminoglykoside im Forschungsfokus. Die Aminoglykoside sind Derivate von klinisch erprobten Antibiotika. Prof. Dr. Timor Bassov vom Technicon in Haifa, Israel, hat sie modifiziert, um eine Verbesserung der Überleseleistung und eine Reduzierung der Toxizität zu erreichen. Ein Designer-Aminoglykosid der ersten Generation wurde von den Mainzer Forschern auch schon früher erfolgreich zum Überlesen von Nonsense-Mutationen in Usher-Genen eingesetzt.
Die Wissenschaftler konnten nun zeigen, dass vor allem durch PTC124 (Ataluren®) und ein Aminoglykosid der zweiten Generation (NB54) das Überlesen des Stopp-Signals im mutierten USH1C-Gen induziert wird. Dadurch kann die Proteinsynthese weiterlaufen und das funktionelle Genprodukt in den Zell- und Organkulturen synthetisiert werden. Insgesamt zeigten die beiden Wirkstoffe PTC124 und NB54 in der Studie neben der verbesserten Überleseeigenschaft auch eine erhöhte Verträglichkeit in Netzhautkulturen der Maus und des Menschen im Vergleich zu klinisch eingesetzten Antibiotika. Zudem gelang es dem Team, das Überlesen der Mutation in Augen von Mäusen in vivo nachzuweisen.
„Unsere genbasierenden Therapiestrategien, die Genreparatur sowie die ‚Read-through‘-Therapie, stellen aus heutiger Sicht wertvolle, vielversprechende Alternativen zur Genaddition mittels Viren dar und dürften vor allem bei den großen und isoformreichen USH-Genen die einzigen Therapieoptionen sein. Wir hoffen, dass wir mit diesen Alternativen einen wertvollen Beitrag zur Therapie nicht nur von Usher-Syndrom-Patienten, sondern auch von weiteren erblich bedingten Retinopathien sowie anderen Erbkrankheiten leisten können“, erklärt Dr. Kerstin Nagel-Wolfrum.
Neben weiterführenden präklinischen Untersuchungen zur Anwendung der Wirkstoffe plant das Mainzer Usher-Labor, das neuartige Verfahren zur Therapie des Usher-Syndroms möglichst zeitnah in die Klinik direkt zum Patienten zu bringen.
Die translationalen biomedizinischen Forschungsarbeiten zur Therapie des Usher-Syndroms wurden mithilfe der Fördermittel des EU-FP7-Projekts „SYSCILIA“, der FAUN-Stiftung und der amerikanischen Foundation Fighting Blindness (FFB) durchgeführt. Die beiden Doktoranden waren Forschungsstudenten und Kollegiaten des durch die Deutsche Forschungsgemeinschaft geförderten Graduiertenkollegs 1044 „Entwicklungsabhängige und krankheitsinduzierte Modifikationen im Nervensystem“. Die Arbeiten der Usher-Forscher sind im Mainzer Forschungsschwerpunkt Translationale Neurowissenschaften (FTN) integriert.
Veröffentlichungen:
Nora Overlack, Tobias Goldmann, Uwe Wolfrum, Kerstin Nagel-Wolfrum
Gene repair of an Usher syndrome causing mutation by zinc-finger nuclease mediated homologous recombination
Investigative Ophthalmology & Visual Science, Juni 2012
DOI: 10.1167/iovs.12-9812
Tobias Goldmann et al.
A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation
EMBO Molecular Medicine, Oktober 2012
DOI: 10.1002/emmm.201201438
Weitere Informationen:
Dr. Kerstin Nagel-Wolfrum
Zell- und Matrixbiologie
Institut für Zoologie
Johannes Gutenberg-Universität Mainz (JGU)
D 55099 Mainz
Tel. +49 6131 39-20131 oder 39-23934
Fax +49 6131 39-23815
E-Mail: nagelwol@uni-mainz.de
http:/www.ag-wolfrum.bio.uni-mainz.de
Media Contact
Weitere Informationen:
http://www.uni-mainz.de/presse/46349.phpAlle Nachrichten aus der Kategorie: Biowissenschaften Chemie
Der innovations-report bietet im Bereich der "Life Sciences" Berichte und Artikel über Anwendungen und wissenschaftliche Erkenntnisse der modernen Biologie, der Chemie und der Humanmedizin.
Unter anderem finden Sie Wissenswertes aus den Teilbereichen: Bakteriologie, Biochemie, Bionik, Bioinformatik, Biophysik, Biotechnologie, Genetik, Geobotanik, Humanbiologie, Meeresbiologie, Mikrobiologie, Molekularbiologie, Zellbiologie, Zoologie, Bioanorganische Chemie, Mikrochemie und Umweltchemie.


Neueste Beiträge

Bakterien für klimaneutrale Chemikalien der Zukunft
Forschende an der ETH Zürich haben Bakterien im Labor so herangezüchtet, dass sie Methanol effizient verwerten können. Jetzt lässt sich der Stoffwechsel dieser Bakterien anzapfen, um wertvolle Produkte herzustellen, die…

Batterien: Heute die Materialien von morgen modellieren
Welche Faktoren bestimmen, wie schnell sich eine Batterie laden lässt? Dieser und weiteren Fragen gehen Forschende am Karlsruher Institut für Technologie (KIT) mit computergestützten Simulationen nach. Mikrostrukturmodelle tragen dazu bei,…

Porosität von Sedimentgestein mit Neutronen untersucht
Forschung am FRM II zu geologischen Lagerstätten. Dauerhafte unterirdische Lagerung von CO2 Poren so klein wie Bakterien Porenmessung mit Neutronen auf den Nanometer genau Ob Sedimentgesteine fossile Kohlenwasserstoffe speichern können…

